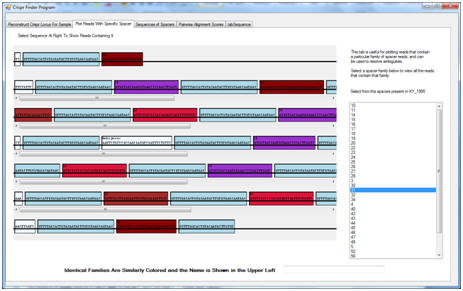
To annotate the CRISPR elements in our 454 data and Illumina data I designed the program CRISPR Finder. This GUI program reconstructs a CRISPR locus from the sequence data, and simultaneously provides visualization tools that allow the user to validate the computational reconstruction, detect polymorphism, and manually check any ambiguities that may appear during the reconstruction.
Briefly, the program works as follows. Each read generated from a sample is checked to see if it contains a sequence similar to the CRISPR repeat present in the ancestral genome. Any reads that contain such a sequence are selected into a subset of reads for further inspection. For each read in this subset, a dynamic alignment algorithm is used to identify the portions of the read that belong to the CRISPR repeats, and by exclusion, those portions that belong to the CRISPR spacers. Spacers that are exactly the same, or that appear to only differ due to sequencing errors, are then identified as spacer families, and these families are each given a numeric name determined by their (essentially random) order of discovery. Finally, as in many modern genome assemblers, the complete CRISPR locus is reconstructed through means of a graph. Each spacer family represents a node that can be placed either upstream or downstream of other families that appear in the same read as themselves. CRISPR Finder constructs this graph, determines the order of the observed spacer families, and plots it in simple format for the user.
Screenshot: Viewing the reads and how they are annotated (identically colored sections represent either repeats or spacers in the same family).
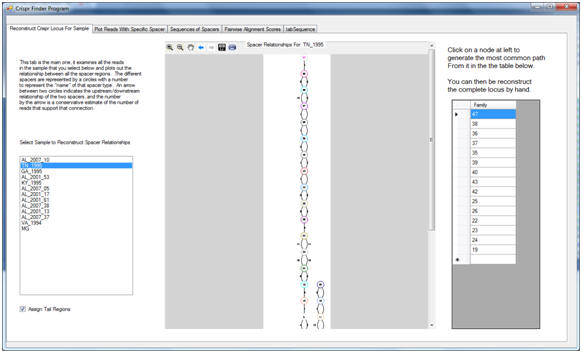
Screenshot 2: Graphical reconstruction of the above CRISPR region, indicating the reads that connect them.