Mycoplasma gallisepticum, House Finches and in situ microbial evolution.
Collaborators: Scott Edwards, Geoff Hill, Camille Bonneaud, Peter Tsai, Allen Rodrigo

© Larry Thompson, Discoverlife.org
Most of my research is conducted by evolving or manipulating bacteria in the lab, which satisfies my proclivity for highly controlled and replicated experiments that support firm conclusions without many assumptions. However, almost nothing in the world evolves under controlled laboratory conditions, and so I am always interested in studies that examine the evolutionary process as it occurs in nature.
I recently was able to study a fantastic model system that allows one to look at the evolution of a microbial population in the wild through time, as it had been sampled from for many years providing a time-series of genetic changes. These samples through time are similar to what I create in my experimental evolution work and have all the same attractive advantages. With a time-series I could estimate evolutionary rates and study genetic changes in a way that simply couldn't be done when one only has samples from only the present day. This is the typical case that past investigators had worked with and it meant all the results were heavily dependent on model assumptions and there were many statistical problems that typically are not accounted for.
The other great advantage of the system is that it has essentially a known founder event that isolated it completeley from it's source population, which makes statements about the evolutionary process even more certain as recombination from an outside population doesn't contaminate the data.
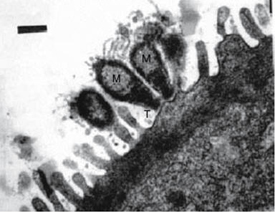
Mycoplasma attached to the epithelium of a chicken where it often resides.
What is this system? Well in 1994 an infection of Mycoplasma gallisepticum (a poultry parasite) was first reported in the House Finch (Carpodacus mexicanus). Shortly after this initial report, the disease began to rapidly spread to other parts of the United States while decimating the Finch populations it came across, with the only upside being that scientists were taking samples of the pathogen while it spread. I examined the genomic changes that occurred in the parasite Mycoplasma population during the epizoodic between 1994 and 2007 by pyrosequencing 12 strains sampled from House Finches throughout this time period as well as Illumina sequencing 4 strains isolated from the source poultry population. The results of this study are in a paper currently under review, but some of the conclusions from it are presented here.
Mycoplasma evolve incredibly fast
The bacteria have long be some of the strangest from a morphological perspective and also some of the fastest evolving bacteria on earth. We found that genetic variation is introduced at a rate of ~1x10-5 per site per year, which is the fastest estimate I know of and also uniquely postions Mycoplasma as one of the best organisms to study in-situ evolution with as its populations evolve faster than others. Concordant with this incredibly high rate, we also found evidence for a very high load of deleterious mutations, begging the next question, how close are they to mutational meltdown and how are they avoiding it?
Phage dynamics are altered after the host shift
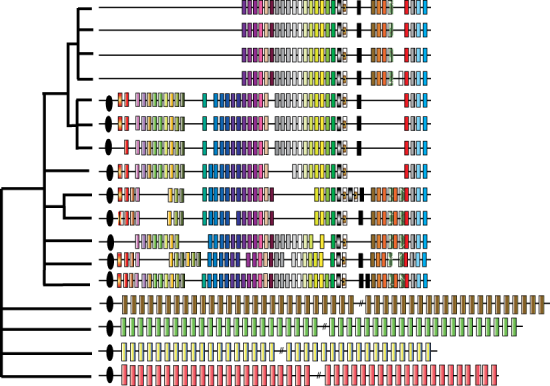
Graphical representation of the reconstructed and aligned CRISPR locus. The bottom four strains samples from the original source population and the top 12 represent samples from the House Finch population, each bar represents a different spacer.
Although long studied as a parasite of poultry, it hasn't been appreciated that M. gallisepticum also has its own parasites, bacteriophages. Examining this genomic data from these populations one of the conclusions I found was that the dynamics of the genomic CRISPR arrays are very different in the original poultry population and the new House Finch population. I wrote a graph-based assembler to reconstruct CRISPR arrays from next generation sequencing data. Using this software we reconstructed the arrays in each strain and found some surprising conclusions. First, CRISPR spacer turnover is extremely rapid in the source population, indicating that phage predation is likely a major component of the population dynamics there. In contrast, following the host shift the CRISPR array effectively goes "silent", no novel spacer elements are present in any of the House Finch MG samples, indicating that the CRISPR array ceased recruiting additional spacers around the time of host switch into the House Finch. During the 13-year period of the epizootic, the number of unique spacers present in the CRISPR array of the samples decreased to 28 and the complete loss of the four CRISPR-associated (i.e. “CAS”) genes occurs by the time of 2007 rendering the entire system non-functional. This could indicate that parasite release, a long observed macro-evolutionary trend, may have contributed to the increased virulence within the House Finch. I also attempted to model the gain and loss of CRISPR spaces to determine there usefulness as phylogenetic markers. Unfortunately, I found that they are not at all useful as their mutational dynamics are too complex to be inferred accurately, though people may still attempt to do so as the single-step model still seems quite popular for microsattellites.
Mutational biases are strong and selective ones are clear
Mycoplasma have a very low GC content and we can ask in this system how much of this could be explained by mutational biases. Within the House Finch Mycoplasma samples, dn/ds values are indistinguishable from one as selection has not had enough time to greatly bias the observed mutations. This means that we can get a great estimate for the underlying mutational frequency, and as shown below this has lead to perpelexing observations which I am currently investigating. I have also shown the IS elements in this genome move around at a very fast rate, but that most insertions do not persist in the population as they are filtered out by selection much faster than SNPs.
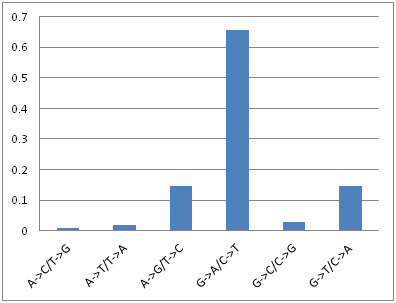
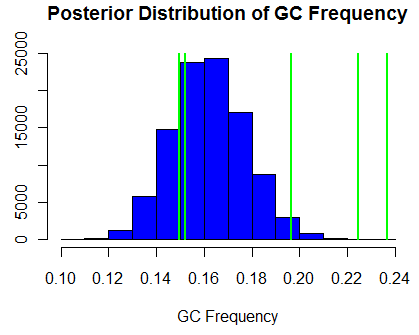
Shown at right are the relative frequencies of different mutations normalized for genomic content derived after polarizing SNPs. I used a uniform dirichelt prior on the mutational types to compute the posterior distribution of equilibrium GC frequency shown at left. This distribution is significantly lower than the 31% observed frequency, indicating some other force maintains the empirically observed frequency. Green lines show the GC frequency at the third position for each of the 5 four-fold degenerate amino acids in the genetic code. This indicates that the calculate GC equilibrium values are likely, but also that other factors affect codon use besides mutational biases as these lines are more seperated than we might expect.
This work has recently been published in PLoS Genetics, and can be found at this link. Unfortunately, the supplements at PLoS must be uploaded as separate files, but a single file with all the supplements (and the annotations next to the figures) is available for download in a combined file here.