C# and .NET are some of the fastest high level languages, but still cannot truly compete with C/C++ for low level speed, and C# code can be anywhere from 20%-300% slower. This is despite the fact that the C# compiler often gets as much information about a method as the C compiler. It has been suggested that SSE/SIMD instructions in C# could overcome some of these issues. To evaluate this, I took a famous computational task, re-coded it using C# SIMD instructions, and evaluated the performance by looking at the execution time and how the emitted assembly code compared to the optimal assembly code.
Background
In 2009, Miguel de Icaza demonstrated a framework that allows C# to use SSE2 intrinsic operations.This is now part of the Mono library and in theory, such methods can greatly save computational time (1.5X-16X), particularly for operations on byte arrays, a common task in bioinformatics.
Test Case
Computing the mandelbrot set is one of the tasks of the computer benchmarks game, which compares program speed in different languages. Currently, C# is listed as being slower than Java, though the programs in each language use different techniques and neither uses an optimal algorithm (see here for a better one). However, it makes a useful test case for raw computing speed. The challenge is to compute the picture shown below by recursively squaring a complex number and adding a constant to it.

![image_thumb[1]](https://i0.wp.com/evolvedmicrobe.com/blogs/wp-content/uploads/2013/09/image_thumb11.png?resize=244%2C237 "image_thumb[1]")
The Algorithm
Complex numbers are a particularly challenging case because their multiplication is not a simple operation, but involves a rather annoyingly squaring of different terms and then adding/subtracting them.

These are not easy vector operations, and as will be shown later one result of this is that using SSE to speed up the values for one multiplication is useless (I tried, it was worse). So, the performance improvement comes from doing two elements of the loop at a time (this is how the current Java submission is faster than the C# one, though it does not use SIMD). The current C# version does the inner loop of the program without SIMD instructions as follows:
|
|
int i = 49; do { zi = zr * zi + zr * zi + ci; zr = tr - ti + cr; tr = zr * zr; ti = zi * zi; } while ((tr + ti <= 4.0) && (--i > 0)); |
This loop is iterating until convergence (<4) or until the max iterations (i<0) have expired. See the wikipedia page for a full explanation. Of interest here is that the “t” variables exist for pipelining purposes, so this is a reasonably optimized code snippet.
Trying to Implement the Optimal Solution Using Mono.Simd
Actually figuring out how to do the complex arthimetic with SSE2 is rather challenging. Fortunately Intel published a document giving the best solution as hand coded assembly, which involves using some special SSE3 instructions I was not aware of. Notably, the Intel optimized code is far faster than even their C code, but is only about 25% better than their optimized assembly code without SSE3.
The code below shows my attempt to implement the Intel approach in Mono. It should be pretty fast, however it may suffer from one fatal flaw. The comparison at the end to check for the final conditions currently requires unpacking both values in the Vector2D (when either Length.X or Length.Y have been < 4.0 at least once, then the loop stops). The comparison for both X and Y can be done in one operation in SIMD using the built in less than statement. However, I do not know how to turn that into a control loop function, as this requires a movmskps assembly function which mono does not seem to expose.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 |
int i = 49; int b = 0; do { Vector2d Zri=Zr*Zi; //calculate r*i for both Zi=Zri+Zri+Ci; //double that and add a constant Zr=Temp; //pre-calculated on previous loop var V0=Zr.InterleaveLow(Zi); //r0,i0 var V1=Zr.InterleaveHigh(Zi); //r1,i1 V0=V0*V0; //r0^2,i0^2 V1=V1*V1; var Length=V0.HorizontalAdd(V1); //(r0^2+i0^2),(r1^2+i1^2) Temp=V0.HorizontalSub(V1)+Cr; //(r0^2-i0^2),(r1^2-i1^2) //now to determine end condition, if (Length.X > 4.0) {b|=2; if (b==3) break;} if (Length.Y>;4.0) {b|=1; if (b==3) break;}} while (--i > 0); |
Results – It’s Faster on Ubuntu

Shown above are the times for three different algorithms. The first is the original best submission on the website. My SIMD submission is shown on the bottom. Because the SIMD version does two cycles within each inner loop, as the Java submission does, I tested the Java submission converted to C# as well. Compared to the current submission this version shows a 81% improvement, but clearly much of that is simply from doing 2 cycles in one loop. However, even once this change is made, the SIMD instructions still give a performance boost.
The Assembly Generated is Still Not Optimal
Although the assembly generated did include lots of SSE commands, in general inspecting the assembly I noticed several things.
-
Never unpack the double array values! My first attempt tried to do the SSE2 steps with those instructions, and then unpack the single values as needed. However, this failed prettty miserably, as accessing the double seemed to involve a lot of stack to register movement.
-
All the XMM registers are not used. The optimal version uses all of them, the C# version uses only 0-3, and soften moves things to and from the stack. Not using registers optimally seems to be a common problem though with C#.
Conclusions
The SIMD version was indeed a fair bit faster, which is nice! However, in this case it was not a game-changer. Most importantly though, it was really easy to use, and I think I might incorporate the byte array operations at some point in future code. This also gave me an appreciation for assembly, which in contrast to what I had heard is easy to read and seems easy to optimize. I just submitted the code to the shoot-out, assuming it works there it should be up soon, and I would love for someone to show me how to fix the end statement.
 The comparison to samtools is based on the system time to parse the file using this task on the
same unix server used for the C# tests.
The comparison to samtools is based on the system time to parse the file using this task on the
same unix server used for the C# tests.

![image_thumb[1]](https://i0.wp.com/evolvedmicrobe.com/blogs/wp-content/uploads/2013/09/image.png "image_thumb[1]")







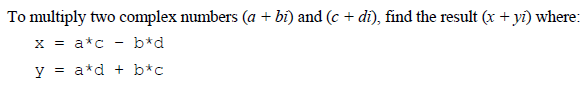{kind=link}